Tools
GitHub Repositories
Selected Software
RawHash, RawHash2 and Rawsamble: RawHash (and RawHash2) is a hash-based mechanism to map raw nanopore signals to a reference genome in real-time. To achieve this, it 1) generates an index from the reference genome and 2) efficiently and accurately maps the raw signals to the reference genome such that it can match the throughput of nanopore sequencing even when analyzing large genomes (e.g., human genome). Rawsamble builds on the same collision‑resistant hashing framework in a reference‑free setting: it performs an all‑vs‑all search for long overlaps directly between raw signals, outputs a PAF overlap graph, and can drive assemblers such as miniasm to generate de novo assemblies—without ever base‑calling the signals.
Below figure shows the overview of the steps that RawHash takes to find matching regions between a reference genome and a raw nanopore signal.
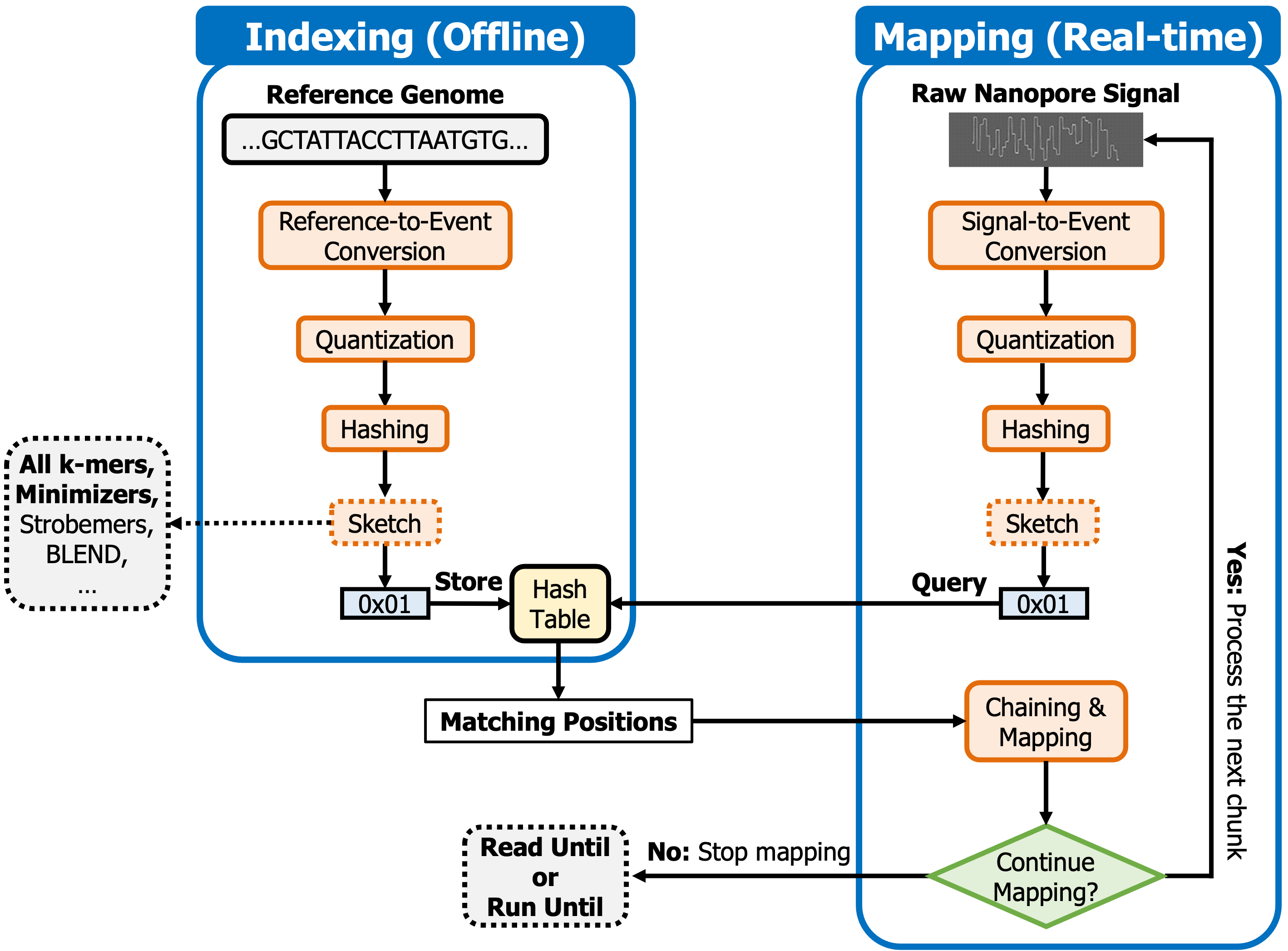
To efficiently identify similarities between a reference genome and reads, RawHash has two steps, similar to regular read mapping tools, 1) indexing and 2) mapping. The indexing step generates hash values from the expected signal representation of a reference genome and stores them in a hash table. In the mapping step, RawHash generates the hash values from raw signals and queries the hash table generated in the indexing step to find seed matches. To map the raw signal to a reference genome, RawHash performs chaining over the seed matches.
RawHash can be used to map reads from FAST5, POD5, SLOW5, or BLOW5 files to a reference genome in sequence format.
RawHash performs real-time mapping of nanopore raw signals. When the prefix of reads can be mapped to a reference genome, RawHash will stop mapping and provide the mapping information in PAF format. We follow the similar PAF template used in UNCALLED and Sigmap to report the mapping information.
GitHub Link | RawHash Paper Link (ISMB/ECCB 2023 Proceedings) | RawHash2 Paper
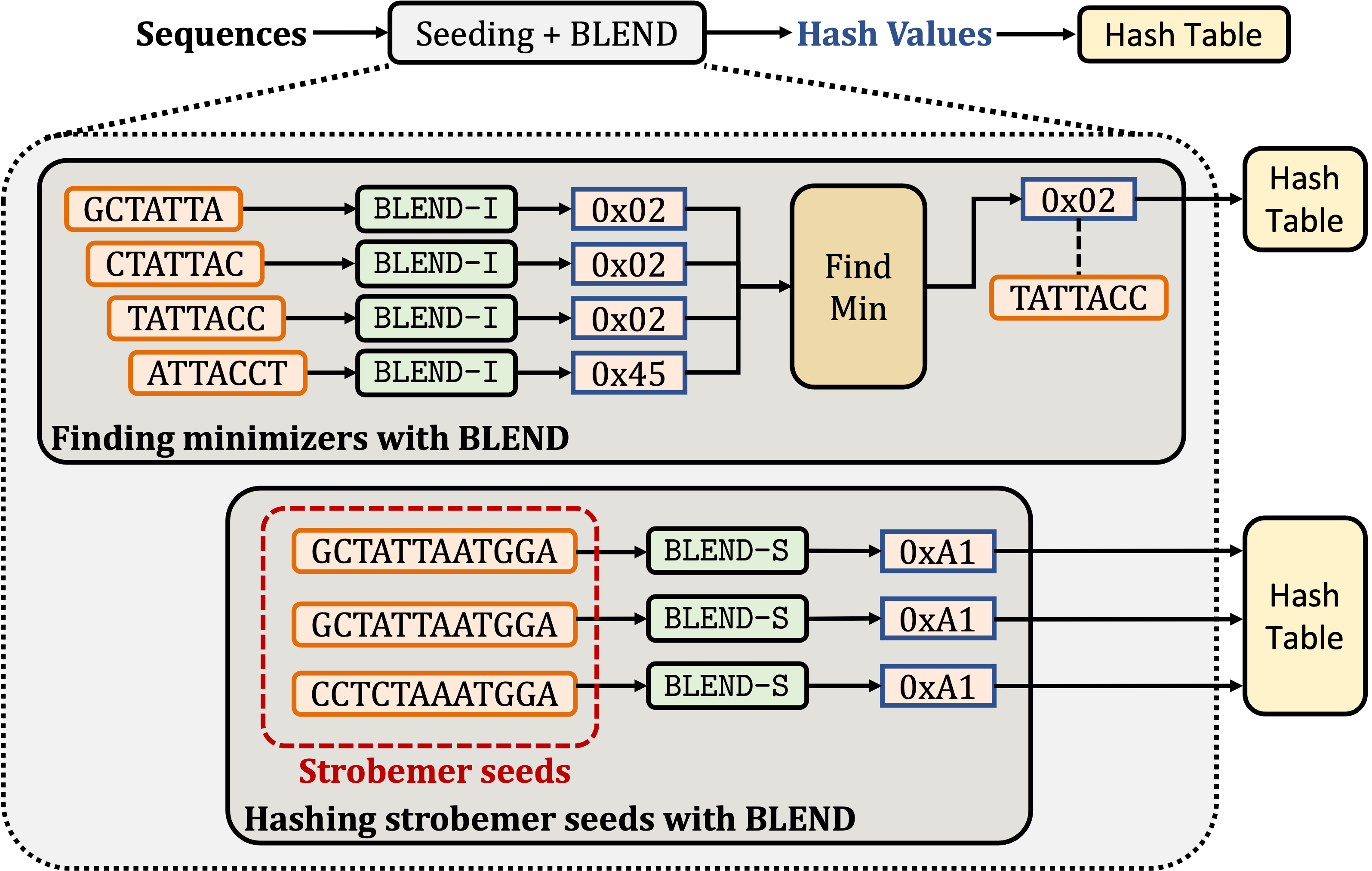
BLEND: BLEND is a mechanism that can efficiently find fuzzy (approximate) seed matches between sequences to significantly improve the performance and accuracy while reducing the memory space usage of two important applications: 1) finding overlapping reads and 2) read mapping. To achieve this, BLEND provides mechanisms to generate the same hash value for highly similar seeds such that BLEND can find fuzzy seed matches with a single lookup from the hash values of seeds. BLEND can be integrated into any seeding mechanism by properly replacing the hash functions of these seeding technqiues. As proof of work, we integrate the BLEND mechanism into minimap2 to generate the hash values for minimizer and strobemer seeds.
GitHub Link | BLEND Paper Link (NARGAB)
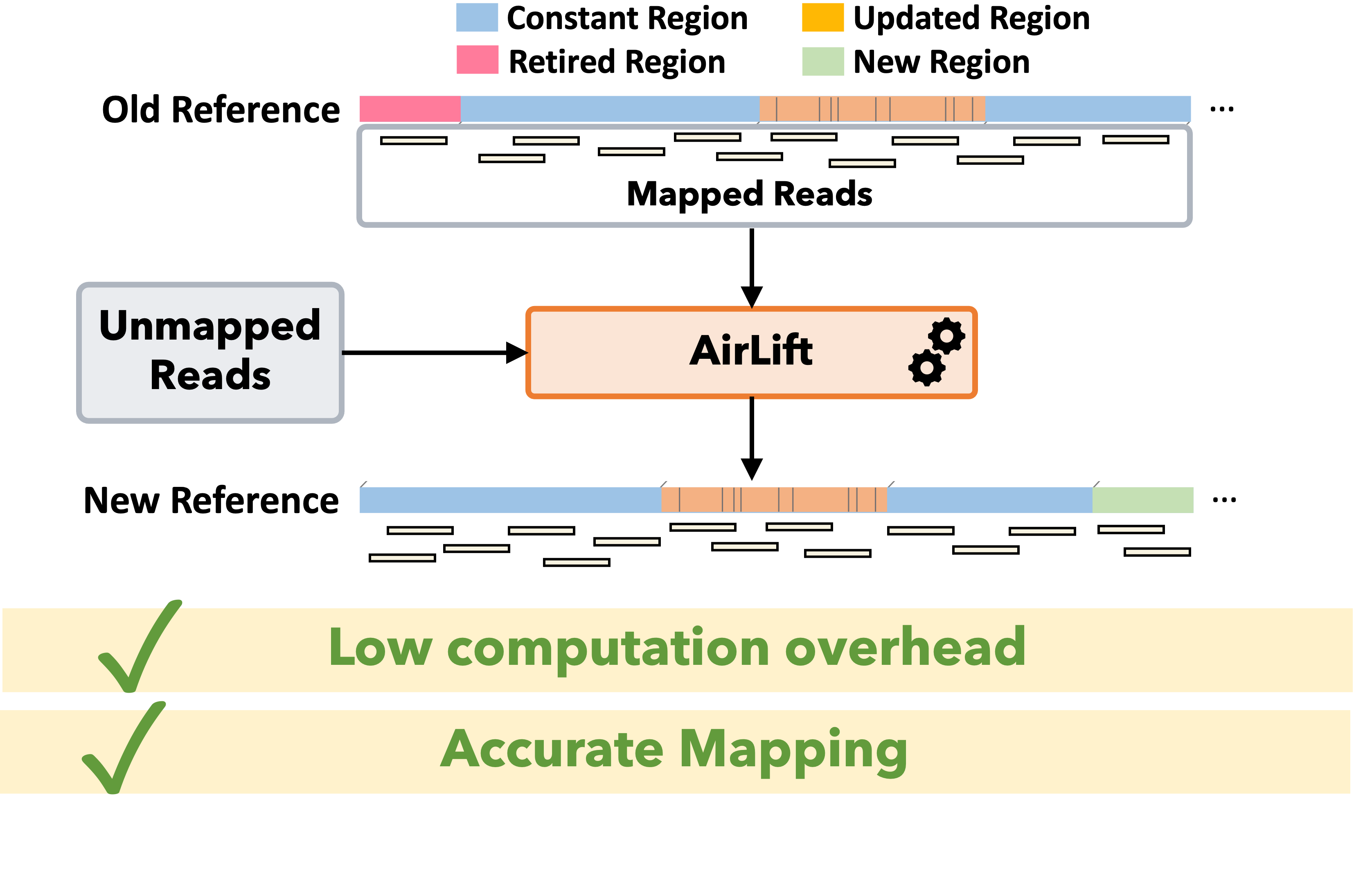
AirLift: As genome sequencing tools and techniques improve, researchers are able to incrementally assemble more accurate reference genomes, which enable sensitivity in read mapping and downstream analysis such as variant calling. A more sensitive downstream analysis is critical for better understanding the health data of a genome donor. Therefore, read sets from sequenced samples should ideally be mapped to the latest available reference genome. Unfortunately, the increasingly large amount of available genomic data makes it prohibitively expensive to fully re-map each read set to its respective reference genome every time the reference is updated. There are several tools that attempt to accelerate the process of updating a read data set from one reference to another (i.e., remapping) by 1) identifying regions that appear similarly between two references and 2) updating the mapping location of reads that map to any of the identified regions in the old reference to the corresponding similar region in the new reference. The main drawback of existing approaches is that if a read maps to a region in the old reference that does not appear similarly in the new reference, the read cannot be remapped. We find that, as a result of this drawback, a significant portion of annotations are lost when using state-of-the-art remapping tools. To address this major limitation in existing tools, we propose AirLift, a fast and comprehensive technique for moving alignments from one genome to another. AirLift reduces 1) the number of reads that need to be fully mapped from the entire read set by up to 99.99% and 2) the overall execution time to remap read sets between two reference genome versions by 19.6x, 6.6x, and 2.7x for large (human), medium (C. elegans), and small (yeast) reference genomes, respectively. We validate our remapping results with GATK and find that AirLift provides similar rates of identifying ground truth SNPs and INDELs as fully mapping a read set.
Apollo: Third-generation sequencing technologies can sequence long reads that contain as many as 2 million base pairs. These long reads are used to construct an assembly (i.e. the subject’s genome), which is further used in downstream genome analysis. Unfortunately, third-generation sequencing technologies have high sequencing error rates and a large proportion of base pairs in these long reads is incorrectly identified. These errors propagate to the assembly and affect the accuracy of genome analysis. Assembly polishing algorithms minimize such error propagation by polishing or fixing errors in the assembly by using information from alignments between reads and the assembly (i.e. read-to-assembly alignment information). However, current assembly polishing algorithms can only polish an assembly using reads from either a certain sequencing technology or a small assembly. Such technology-dependency and assembly-size dependency require researchers to (i) run multiple polishing algorithms and (ii) use small chunks of a large genome to use all available readsets and polish large genomes, respectively.We introduce Apollo, a universal assembly polishing algorithm that scales well to polish an assembly of any size (i.e. both large and small genomes) using reads from all sequencing technologies (i.e. second- and third-generation). Our goal is to provide a single algorithm that uses read sets from all available sequencing technologies to improve the accuracy of assembly polishing and that can polish large genomes. Apollo (i) models an assembly as a profile hidden Markov model (pHMM), (ii) uses read-to-assembly alignment to train the pHMM with the Baum-Welch algorithm and (iii) decodes the trained model with the Viterbi algorithm to produce a polished assembly. Our experiments with real readsets demonstrate that Apollo is the only algorithm that (i) uses reads from any sequencing technology within a single run and (ii) scales well to polish large assemblies without splitting the assembly into multiple parts.
GitHub Link | Apollo Paper Link (Bioinformatics)
ApHMM‑GPU: ApHMM‑GPU is the first GPU implementation of the Baum–Welch expectation‑maximization algorithm for profile Hidden Markov Models (pHMMs). It accelerates the forward–backward and parameter‑update steps across thousands of CUDA cores, delivering large speed‑ups over CPU baselines and enabling fast, energy‑efficient genome analysis. GitHub Link
Hercules: Hercules applies profile Hidden Markov Model‑based statistical learning to correct systematic sequencing errors in both DNA and RNA long‑read datasets, boosting the accuracy of downstream mapping and assembly.
GitHub Link
On Genomic Repeats and Reproducibility: This repository contains scripts and pipelines that detect, quantify, and report reproducibility issues in popular variant‑calling workflows (e.g., BWA‑MEM + GATK HaplotypeCaller) that stem from repetitive genomic regions, helping researchers build more robust analyses.
GitHub Link